
Simulaciones
Protocolos para simulaciones de dinámica molecular de última generación
Simule sistemas biológicos con las mejores herramientas de su clase
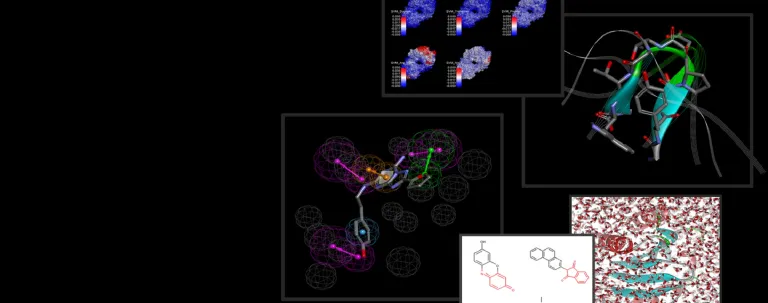
Los procesos biomoleculares se basan en una variedad de interacciones dinámicas entre proteínas, ligandos, disolventes e iones. A menudo, los detalles específicos de estas interacciones son difíciles de captar a través de la experimentación física solamente debido a las escalas de tiempo cortas en las que se producen. La simulación puede ayudar a determinar la energía de estos procesos, proporcionando información sobre su mecanismo de acción y sus propiedades.
BIOVIA Discovery Studio utiliza los mejores programas de simulación molecular de su clase, NAMD y CHARMm. Además, la dinámica molecular acelerada gaussiana (GaMD) también se ha implementado en la última versión de Discovery Studio para el muestreo mejorado simultáneo y sin restricciones y los cálculos de energía libre.
- Simulación
- Modelo
- Explorar
Simulación
- CHARMm
- NAMD
- Realice simulaciones de dinámica molecular basadas en disolventes explícitos.
- Realice la solvatación de una proteína con membrana explícita y ejecute simulaciones de MD.
- DMol3/CHARMm
- Calcule energías de punto único o realice minimizaciones de complejos receptor-ligando mediante simulaciones híbridas de mecánica cuántica/mecánica molecular (QM/MM).
- Implementación de GaMD para un muestreo mejorado sin restricciones y cálculos de energía libre simultáneos.
- Configure y ejecute un equilibrado de GaMD, parametrizando automáticamente los potenciales de refuerzo necesarios.
- Ejecute y reinicie simulaciones de GaMD.
- Calcule un panorama de energía libre a partir de un conjunto de trayectorias de MD, permitiendo la reponderación estadística de simulaciones de GaMD.
Modelo
- Compatibilidad con una amplia variedad de campos de fuerza, incluidos CGenFF, charmm36, CHARMm, etc.
- Método MATCH para tipificar ligandos con charmm36
- Soporte completo del mecanismo de aplicación de parches de CHARMM
- Método de solvatación acuosa explícita rápida con contraiones opcionales, adecuado para sistemas moleculares muy grandes
- Solvatación de proteína transmembrana en una bicapa lipídica preequilibrada
- Análisis de las trayectorias de MD
Explorar
- Realice predicciones rápidas y precisas de ionización de proteínas y pKs de residuos para la preparación de proteínas.
- Utilice CDOCKER, un motor de acoplamiento basado en CHARMm, para realizar un acoplamiento y refinamiento flexibles basados en ligandos.
- Optimice la conformación de múltiples ligandos en el contexto de un receptor.
- Calcule las energías de enlace de las conformaciones acopladas.
- Pronostique con precisión la energía relativa de unión de ligandos para una serie de ligandos congenéricos utilizando el método de perturbación de energía libre (FEP).
- Calcule la energía libre relativa de unión para una biblioteca combinatoria de ligandos modelada mediante Multi-Site Lambda Dynamics (MSLD).
- Calcule la energía libre de unión de ligandos y estudie su desacoplamiento mediante simulaciones de dinámica molecular dirigida (SMD) basadas en CHARMm.
- Examine los efectos del potencial electrostático con la ecuación de Poisson-Boltzmann (PB) basada en CHARMm.
Comience su viaje
Acelere el descubrimiento de fármacos con BIOVIA Discovery Studio.
Únase a la conversación en la comunidad de descubrimiento y desarrollo de fármacos de BIOVIA.
Preguntas frecuentes sobre software y programas de dinámica molecular
Una simulación de dinámica molecular (MD) es un método informático que se utiliza para estudiar los movimientos físicos de los átomos y las moléculas a lo largo del tiempo. Ayuda a comprender el comportamiento de sistemas complejos mediante la simulación de interacciones a nivel atómico en determinadas condiciones.
Para ser más precisos, el método de simulación de dinámica molecular (MD) es una técnica computacional utilizada para modelar los movimientos físicos de los átomos y las moléculas a lo largo del tiempo. Simula las interacciones entre partículas (como átomos, moléculas o iones) basándose en la mecánica clásica, utilizando normalmente las leyes de movimiento de Newton. Al calcular las fuerzas entre estas partículas, las simulaciones de MD predicen cómo evolucionan sus posiciones con el tiempo, lo que permite a los investigadores estudiar el comportamiento dinámico de sistemas moleculares complejos, como proteínas, ácidos nucleicos o materiales en diferentes entornos.
Las simulaciones de dinámica molecular proporcionan información sobre cómo interactúan los fármacos con sus proteínas objetivo a nivel atómico. Esto permite a los investigadores predecir afinidades de unión, optimizar los candidatos a fármacos y diseñar terapias más eficaces sin necesidad de una amplia experimentación física.
Las simulaciones de MD ayudan a los investigadores a visualizar las interacciones fármaco-objetivo, a predecir la estabilidad de la unión del fármaco y a evaluar cómo afectan los cambios moleculares a la eficacia de un fármaco. Esto acelera la identificación de posibles candidatos a medicamentos y reduce el tiempo y el coste de los ensayos experimentales.
Existen varios tipos de simulaciones de MD, entre los que se incluyen:
- MD clásica (simulación estándar de sistemas moleculares)
- MD acelerada (para un muestreo más rápido de las conformaciones)
- Mecánica cuántica/mecánica molecular (QM/MM) (simulaciones híbridas para reacciones químicas)
- MD acelerada gaussiana (GaMD), que mejora el muestreo para sistemas biomoleculares complejos.
A continuación se presenta una tabla comparativa que destaca las principales diferencias entre NAMD y CHARMm en las simulaciones clásicas de dinámica molecular, que cubre aspectos como la funcionalidad, la eficiencia computacional, el tamaño del sistema y los casos de uso:
| Característica | NAMD | CHARMm |
|---|---|---|
| Función principal | Especializado en simulaciones paralelas de dinámica molecular para grandes sistemas. | Software de simulación de dinámica molecular centrado en cálculos de campos de fuerza. |
| Desarrollador | Desarrollado por el Grupo de Biofísica Teórica y Computacional (TCBG). | Desarrollado por Accelrys/BIOVIA. |
| Eficiencia computacional | Altamente optimizado para el procesamiento paralelo en clústeres y supercomputadoras. | Eficiente para sistemas más pequeños; menor atención a la paralelización a gran escala. |
| Campos de fuerza soportados | Utiliza principalmente campos de fuerza CHARMM, pero también admite AMBER (programa de simulación biomolecular) y otros. | Basado en el campo de fuerza CHARMM (Chemistry at Harvard Molecular Mechanics). |
| Tamaño del sistema | Adecuado para grandes sistemas biomoleculares, como proteínas y membranas. | Ideal para sistemas más pequeños y simulaciones de QM/MM. |
| Soporte de GPU | Amplio soporte de GPU para la aceleración mediante CUDA. | Aceleración de GPU limitada a través de DOMDEC y OpenMM. |
| Funciones avanzadas | Soporte de simulaciones avanzadas, como MD dirigida, MD de intercambio de réplicas. | Gran compatibilidad con simulaciones de QM/MM y acoplamiento de ligandos. |
| Casos de uso | Ideal para simulaciones a gran escala y de alto rendimiento de proteínas y membranas. | Se utiliza para el diseño de fármacos, el acoplamiento de ligandos y el análisis detallado de campos de fuerza. |
| Facilidad de uso | Requiere cierta complejidad de configuración para la computación paralela. | Más fácil de configurar para sistemas más pequeños o cuando se integra en Discovery Studio. |
| Soporte de la comunidad | Ampliamente utilizado en la investigación académica con una gran comunidad de usuarios. | Mayor nicho, utilizado a menudo en el sector para el descubrimiento de fármacos. |
Las simulaciones de dinámica molecular con BIOVIA Discovery Studio proporcionan potentes herramientas, como NAMD y CHARMm, para modelar con precisión interacciones biomoleculares complejas. Con la MD acelerada gaussiana (GaMD) para un muestreo mejorado y cálculos de energía libre, los investigadores pueden explorar de forma eficaz los procesos moleculares. El software admite varios campos de fuerza y técnicas de modelado molecular, lo que lo hace esencial para el descubrimiento de fármacos, al ayudar a los usuarios a simular, modelar y analizar interacciones a nivel atómico.
Descubra también







Descubra lo que BIOVIA puede hacer por usted
Hable con un experto de BIOVIA para descubrir cómo nuestras soluciones permiten colaborar sin problemas e innovar de manera sostenible en organizaciones de todos los tamaños.
Ponerse en marcha
Los cursos y las clases están disponibles para estudiantes, instituciones académicas, profesionales y empresas. Encuentre la formación de BIOVIA adecuada para usted.
Obtener ayuda
Encuentre información sobre certificación de software y hardware, descargas de software, documentación del usuario, contacto con soporte y oferta de servicios