
仿真
先进分子动力学仿真协议
使用一流的工具对生物系统进行仿真
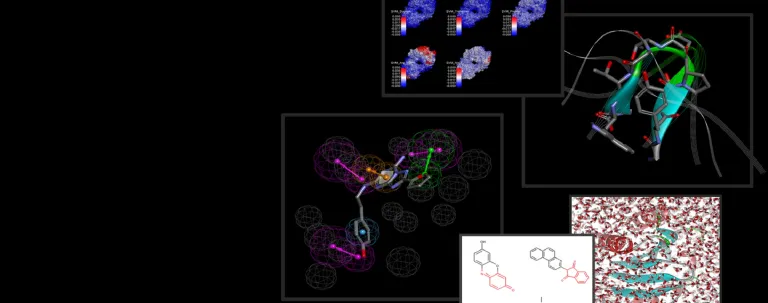
生物分子过程依赖于蛋白质、配体、溶剂和离子之间的各种动态相互作用。通常,由于这些相互作用发生的时间尺度较短,仅通过物理实验很难捕获到具体细节。仿真有助于解释这些流程的能量学原理,从而提供关于其作用机制和特性的深度见解。
BIOVIA Discovery Studio 采用一流的分子仿真程序 NAMD 和 CHARMm。 此外,最新版本的 Discovery Studio 还实施了高斯加速分子动力学 (GaMD) 方法,可同时进行无约束增强采样和自由能计算。
- 仿真
- 建模
- 探索
建模
- 支持多种力场,包括 CGenFF、charmm36、CHARMm 等
- 使用 charmm36 进行配体分型的 MATCH 方法
- 完全支持 CHARMM 补丁机制
- 含可选反离子的快速显式水化方法,适用于非常大的分子系统
- 将跨膜蛋白溶解于预平衡的脂质双分子层中
- MD 轨迹分析
探索
- 执行快速准确的蛋白质电离和残基 pKs 预测,以进行蛋白质制备
- 使用基于 CHARMm 的对接引擎 CDOCKER 进行灵活的基于配体的对接和细化
- 在受体背景下对多个配体进行位姿优化
- 计算对接位姿的结合能
- 使用自由能微扰法 (FEP) 准确预测同类配体系列的相对配体结合能
- 计算根据多位点 λ 动力学 (MSLD) 建模的配体组合库的相对结合自由能
- 使用基于 CHARMm 的拉伸分子动力学 (SMD) 仿真来估计配体结合自由能并研究配体解离
- 使用 CHARMm Poisson-Boltzmann (PB) 方程检查静电势效应
开启您的旅程
利用 BIOVIA Discovery Studio 加速药物发现。
请进入 BIOVIA 药物发现与开发社区,参加精彩的讨论!
关于分子动力学软件和程序的常见问题答疑
分子动力学 (MD) 仿真是一种基于计算机的方法,可用于研究原子和分子随时间变化的物理运动。它通过在给定条件下对原子级相互作用进行仿真,来帮助理解复杂系统的行为。
更准确地说,分子动力学 (MD) 仿真方法是一种用于对原子和分子随时间变化的物理运动进行建模的计算技术。它通常使用牛顿运动定律,基于经典力学对粒子(如原子、分子或离子)之间的相互作用进行仿真。通过计算这些粒子之间的力,MD 仿真可以预测它们的位置如何随时间而变化,从而使研究人员能够研究复杂分子系统(如蛋白质、核酸或材料)在不同环境中的动力学行为。
分子动力学仿真可以帮助我们提供关于药物在原子级上如何与靶蛋白相互作用的深度见解。这样,研究人员无需进行大量的物理实验,即可预测结合亲和力、优化备选药物并设计更有效的疗法。
MD 仿真可帮助研究人员可视化药物-靶标的相互作用,预测药物结合的稳定性,并评估分子变化如何影响药效。这不仅能加速识别有希望的备选药物,还能减少实验性试验的时间和成本。
MD 仿真有多种类型,包括:
- 经典 MD(分子系统的标准仿真),
- 加速 MD(更快速的构象采样),
- 量子力学/分子力学 (QM/MM)(化学反应的混合仿真),
- 高斯加速 MD (GaMD),增强了复杂生物分子系统的采样。
下面的对比表中突出显示了经典分子动力学仿真中的 NAMD 与 CHARMm 之间的主要区别,涵盖功能、计算效率、系统规模和用例等方面:
| 特征 | NAMD | CHARMm |
|---|---|---|
| 主要功能 | 专门处理大型系统的并行分子动力学仿真。 | 专注于力场计算的分子动力学仿真软件。 |
| 开发商 | 由 Theoretical and Computational Biophysics Group (TCBG) 开发。 | 由 Accelrys/BIOVIA 开发。 |
| 计算效率 | 针对集群和超级计算机上的并行处理进行了高度优化。 | 处理较小的系统时效率较高;不太注重大规模并行化。 |
| 支持的力场 | 主要使用 CHARMM 力场,但支持 AMBER(生物分子仿真程序)和其他程序。 | 基于 CHARMM (Chemistry at Harvard Molecular Mechanics) 力场。 |
| 系统规模 | 适用于大型生物分子系统,如蛋白质和膜。 | 适用于较小型系统和 QM/MM 仿真。 |
| GPU 支持 | 广泛的 GPU 支持,可使用 CUDA 加速。 | 通过 DOMDEC 和 OpenMM 获得有限的 GPU 加速。 |
| 先进功能 | 支持先进仿真,如拉伸 MD、副本交换 MD。 | 对 QM/MM 仿真和配体对接提供强大支持。 |
| 用例 | 最适合用于大规模、高性能的蛋白质和膜仿真。 | 用于药物设计、配体对接和详细的力场分析。 |
| 易用性 | 有一定的设置复杂性,适用于并行计算。 | 更易于设置,适用于较小型系统或当集成到 Discovery Studio 中时。 |
| 社区支持 | 广泛应用于学术研究,拥有庞大的用户社区。 | 更小众,常用于行业内的药物发现。 |
使用 BIOVIA Discovery Studio 的分子动力学仿真提供了 NAMD 和 CHARMm 等强大的工具,可以为复杂的生物分子相互作用进行准确建模。借助高斯加速 MD (GaMD) 增强采样和自由能计算,研究人员可以高效地探索分子过程。该软件支持多种力场和分子建模技术,在药物发现中发挥着重要作用,可帮助用户对原子级的相互作用进行仿真、建模和分析。
了解更多







了解 BIOVIA 可以为您做些什么
与 BIOVIA 专家进行交谈,了解我们的解决方案如何在各种规模的企业中实现无缝协作和可持续创新。
了解更多内容
学生、学术界人士、专业人员和企业人员均可参加相关课程并加入班级。查找适合您的 BIOVIA 培训。
获取帮助
查找有关软硬件认证、软件下载、用户文档、支持联系人和服务项目的信息