
BIOVIA TURBOMOLE
高速かつ堅牢な量子化学
分子および結晶のモデリングおよびシミュレーション
量子化学計算は、世界中の研究者にとってなくてはならないものです。カールスルーエ大学およびカールスルーエ研究センター(1989 ~ 2007 年)および TURBOMOLE GmbH (2007 年以降)が共同開発した TURBOMOLE® は、幅広い用途に対応する、第一原理電子状態計算用のための強力で汎用的な量子化学プログラム・パッケージです。
BIOVIA TURBOMOLE は、すべての標準的および最先端の手法、DFT 法または結合クラスター法を使用した分子および固体、励起状態およびスペクトル用の高速な DFT コードを搭載しています。高精度の計算および高速かつ低スケーリングのアルゴリズムと並列化によって、以前は不可能だった系の計算に対応します。
主なメリット
- 結果を導き出すまでの時間を最大 80% 短縮
- 研究者の生産性を 25% 向上
- 物理法則に基づく量子化学計算手法
- 最高水準かつ最先端のツール
- 専門家も専門家以外も操作が可能な強力なツール
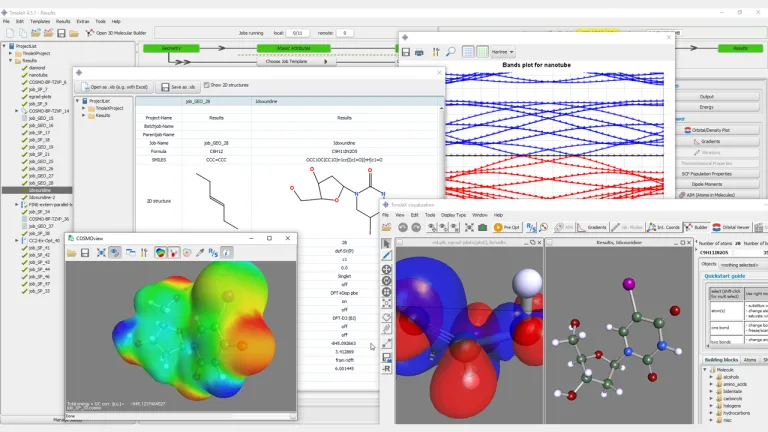
BIOVIA TURBOMOLE は、分子、クラスター、周期系、および溶液の量子化学シミュレーション用の超効率的で安定したツールを提供します。ランダム位相近似(RPA)を含む密度汎関数理論(DFT)、GW-Bethe–Salpeter 法、Møller–Plesset 理論、露に相関した結合クラスター法など、優れた精度コスト比を持つ手法を専門にしています。
- 反応
- 分光法
- 光学装置
- 使いやすさ
反応
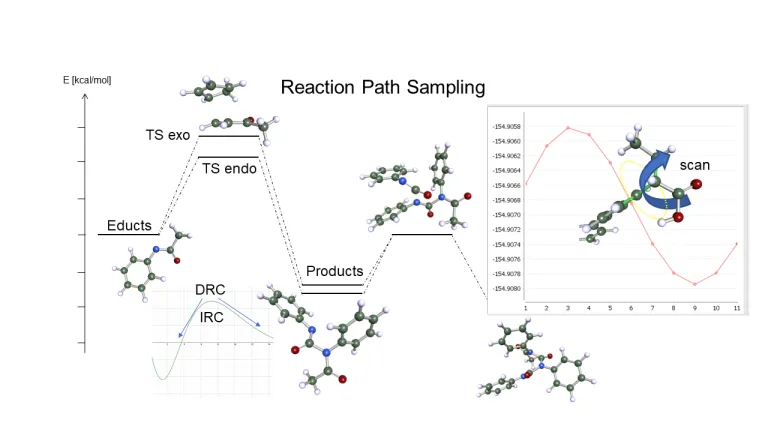
量子化学の主な用途の 1 つは、化学反応の正確な予測と最適化です。溶液中における触媒の最適化などのタスクは、あらゆる業界の科学者にとって挑戦です。さまざまな手法と計算手順を組み合わせる必要がありますが、Turbomole には、クラス最高の精度を達成するために、以下のさまざまなツールと手法が用意されています。
- 反応経路の最適化、遷移状態の探索、ポテンシャル・エネルギー曲面のスキャン
- CCSD (T)や RPA などのゴールド・スタンダードを使用した、非常に正確かつ迅速なエネルギー
- COSMO-RS を使用した比類のない精度の溶媒和効果
- マルチコア・ワークステーションやクラスターなどの標準ハードウェア向けに高度に最適化
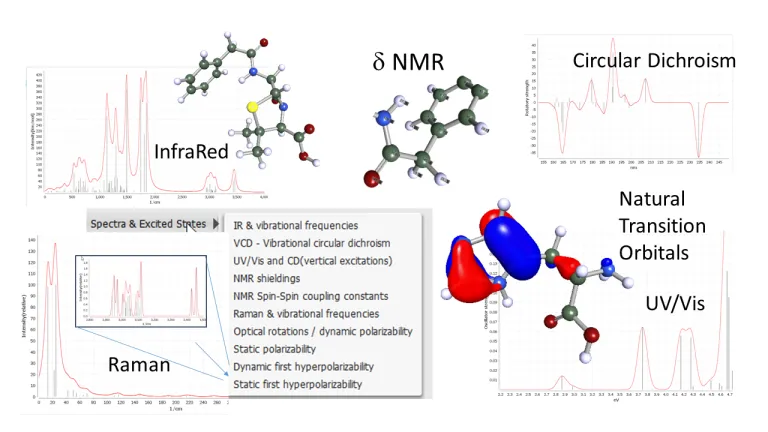
分光法
分光法は、あらゆる分野における構造解析や品質管理に欠かせません。BIOVIA TURBOMOLE では、振動分光法だけでなく光学分光法も含め、以下のようなさまざまな方法によるスペクトルの予測と解析を実行できます。
- 赤外(IR)およびラマン・スペクトル
- 振動円二色性(VCD)
- 紫外可視分光法(紫外および可視スペクトルにおける吸収)、CD (電子円二色性)
- 振動吸収および発光(SVL および VIPER)スペクトル
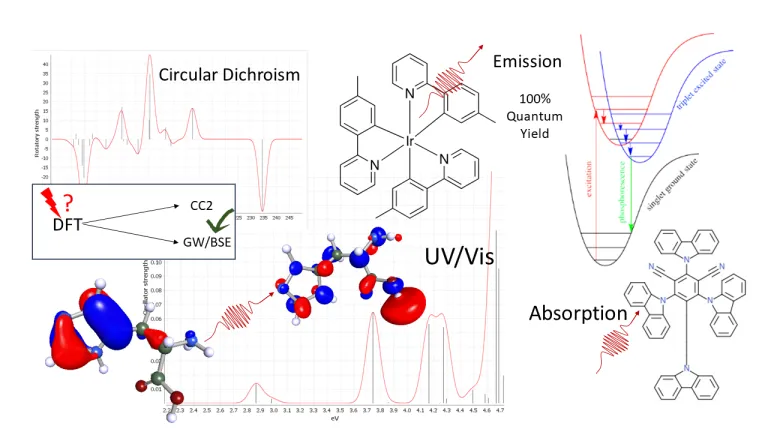
光学装置
分子有機半導体は、量子化学ベースのさまざまな手法を用いてシミュレーションし、発光分子または吸収分子の特性を予測します。BIOVIA TURBOMOLE は、再組織化エネルギー、酸化/還元電位、ギブス自由エネルギーのような特性を自動化されたワークフローという形で提供します。
TURBOMOLE には他に先駆けて軽元素用にスピン軌道カップリング法が導入され、厳密な相対論的全電子計算、次世代 DFT 汎関数、高精度 GW および結合クラスター法を使用します。これにより、電荷移動励起などの一般的な DFT の問題の発生を回避します。
使いやすさ
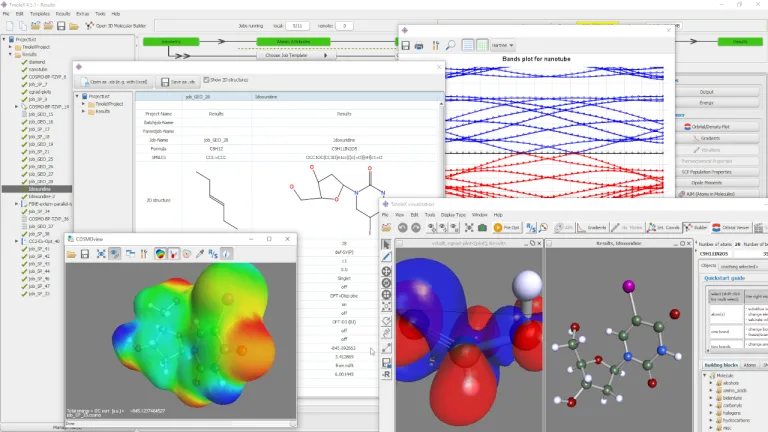
TmoleX は、TURBOMOLE の計算をすばやく処理するための使いやすいグラフィカル・ユーザー・インターフェースです。従来の量子化学スイートはコマンド・ラインやスクリプトを使用するパワー・ユーザー向けのものでしたが、TmoleX なら量子化学の計算処理を簡単に習得して実行できます。TURBOMOLE の使用頻度が高くない場合に最適なツールで、以下が可能です。
- 入力生成と結果の視覚化
- 3D および 2D 分子ビルダー
- さまざまなインポートおよびエクスポート機能
- 多数の手法と特性
さあ、始めましょう
量子化学の世界は変化しています。BIOVIA で一歩先を行く方法を発見しましょう。
その他の情報


BIOVIA の活用方法
組織の規模の大小を問わず、シームレスなコラボレーションと持続可能なイノベーションに、このソリューションがどう役立つかについて、BIOVIA の担当技術者がご説明します。
始めましょう
学生、教育機関、専門家、企業向けのコースとクラスをご用意しています。お客様に最適な BIOVIA トレーニングを受講してください。
サポートを受ける
ソフトウェアやハードウェアの資格認定、ソフトウェアのダウンロード、ユーザー・マニュアル、サポート連絡先、サービス・オファリングに関する情報を入手できます。