
BIOVIA TURBOMOLE
Química cuántica rápida y sólida
Modelado y simulación de moléculas y cristales
Para los investigadores de todo el mundo, los cálculos de química cuántica son indispensables. Con TURBOMOLE®, un desarrollo de la Universidad de Karlsruhe y Forschungszentrum Karlsruhe GmbH (1989-2007) y TURBOMOLE GmbH (desde 2007), ofrecemos un paquete de programa de química cuántica muy potente y de uso general para cálculos de estructuras electrónicas ab initio con una amplia gama de aplicaciones.
BIOVIA TURBOMOLE incluye todos los métodos estándar y de última generación, un código DFT muy rápido para moléculas y sólidos, estados excitados y espectros que utilizan métodos DFT o de clúster acoplado. Los cálculos de alta precisión, así como algoritmos rápidos y de baja escala, y la paralelización permiten abarcar sistemas que antes estaban fuera de su alcance.
Principales ventajas
- Resultados hasta un 80 % más rápidos
- Mejora del 25 % en la productividad de los investigadores
- Métodos de química cuántica computacional basados en la física
- Herramienta tanto de vanguardia como de última tecnología
- Potentes herramientas con las que pueden trabajar expertos y no expertos
BIOVIA TURBOMOLE proporciona herramientas ultraeficientes y estables para simulaciones de química cuántica de moléculas, clústeres, sistemas periódicos y soluciones. Especializado en métodos con una relación precisión-coste excepcional, como la teoría funcional de densidad (DFT), incluida la aproximación de fase aleatoria (RPA), los métodos GW-Bethe-Salpeter, la teoría Møller-Plesset y los métodos de clúster acoplado explícitamente correlacionados.
- Reacciones
- Espectroscopia
- Dispositivos ópticos
- Fácil de usar
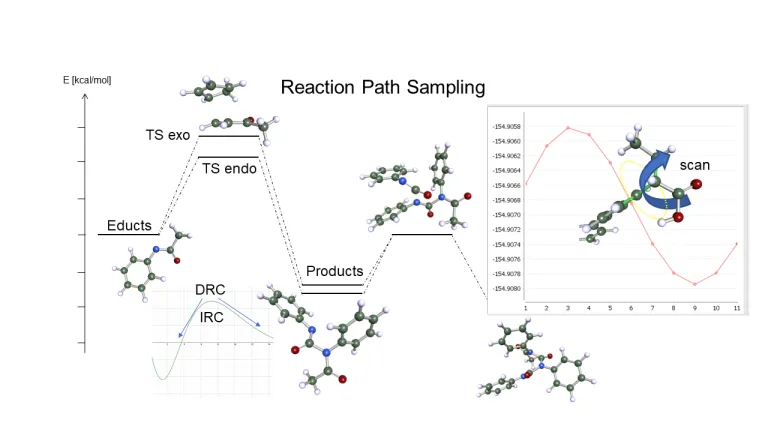
Reacciones
Una de las principales aplicaciones de la química cuántica es la predicción exacta y la optimización de las reacciones químicas. Tareas como la optimización de catalizadores en entornos líquidos son un reto para los científicos de todos los sectores. Se requiere una combinación de diferentes métodos y pasos computacionales y Turbomole ofrece una amplia variedad de herramientas y métodos para lograr la mejor precisión de su clase:
- Optimizaciones de la ruta de reacción, búsquedas de estado de transición, escaneo de superficies de energía potencial
- Energías altamente precisas pero rápidas que utilizan estándares como CCSD(T) o RPA
- Efectos de solvación con una precisión inigualable gracias a COSMO-RS
- Altamente optimizado para hardware estándar, como estaciones de trabajo o clústeres multinúcleo
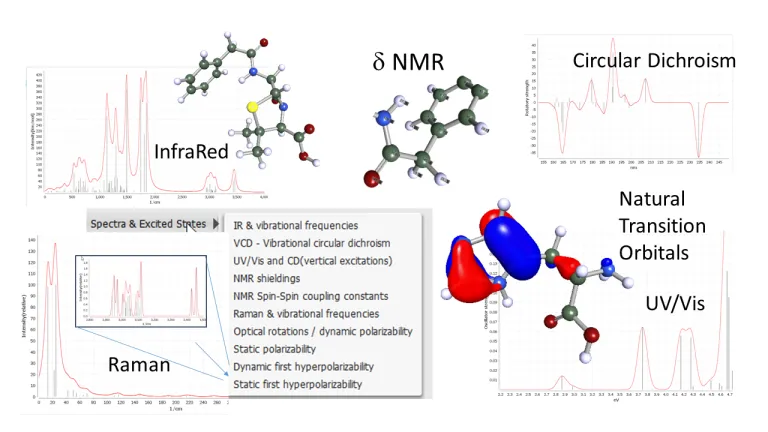
Espectroscopia
La espectroscopia es indispensable para el análisis de estructuras y el control de calidad en todos los campos de aplicación. Con BIOVIA TURBOMOLE, se pueden realizar predicciones y análisis de varios métodos, tanto para espectroscopia vibratoria como óptica:
- Espectros de infrarrojos (IR) y RAMAN
- Dicroísmo circular vibracional (VCD)
- UV-Vis óptico (absorción en espectros UV y visibles), CD (dicroísmo circular electrónico)
- Espectros de emisión y absorción vibrónica (SVL y VIPER)
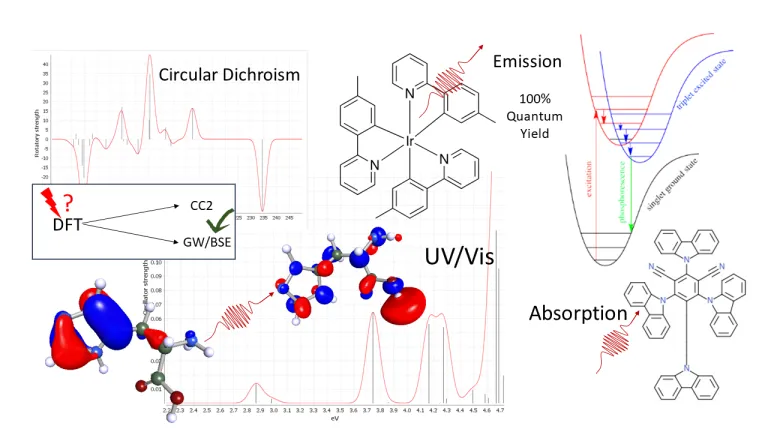
Dispositivos ópticos
Los semiconductores orgánicos moleculares se pueden simular utilizando varios métodos basados en química cuántica para predecir las propiedades de las moléculas que emiten o absorben luz. BIOVIA TURBOMOLE ofrece en un flujo de trabajo automatizado propiedades como energías de reorganización, potenciales de oxidación/reducción o energía libre de Gibbs de solvación.
Los métodos de acoplamiento espín-órbita han sido pioneros en TURBOMOLE para elementos ligeros que utilizan cálculos relativistas exactos de todos los electrones, funciones DFT de última generación y GW de alta precisión, así como métodos de clúster acoplado que no sufren fallos típicos de DFT como excitaciones de transferencia de carga.
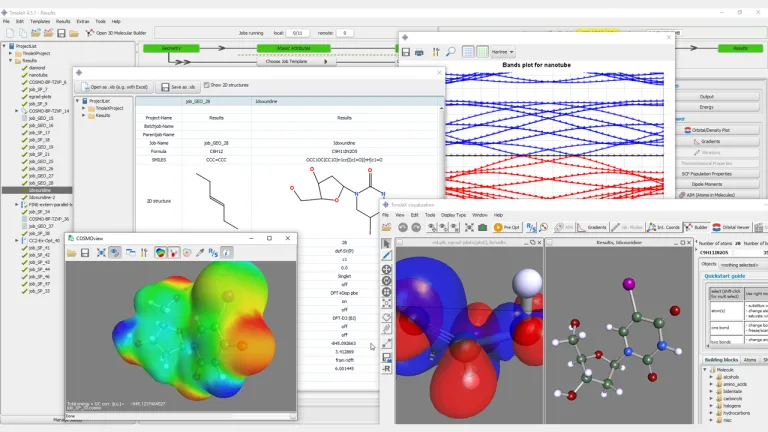
Fácil de usar
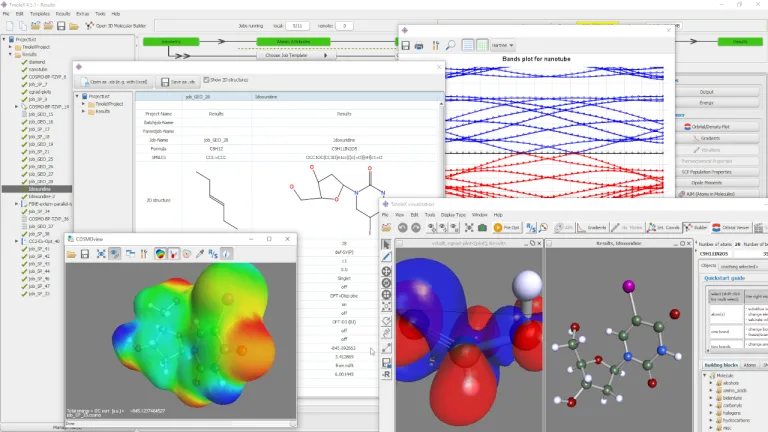
TmoleX es nuestra interfaz gráfica de usuario fácil de usar para gestionar rápidamente los cálculos de TURBOMOLE. Aunque tradicionalmente se han desarrollado conjuntos de química cuántica para un uso basado en línea de comandos o secuencias de comandos (para usuarios avanzados), TmoleX le permite utilizar química cuántica después de unos minutos de introducción. Es la herramienta perfecta para uso ocasional de TURBOMOLE que permite:
- Generación de entradas y visualizaciones de resultados
- Constructores moleculares 3D y 2D
- Varias funciones de importación y exportación
- Un gran número de métodos y propiedades
Comience su viaje
El mundo de la química cuántica está cambiando. Descubra cómo mantenerse un paso por delante con BIOVIA.
Descubra también


Descubra lo que BIOVIA puede hacer por usted
Hable con un experto de BIOVIA para descubrir cómo nuestras soluciones permiten colaborar sin problemas e innovar de manera sostenible en organizaciones de todos los tamaños.
Ponerse en marcha
Los cursos y las clases están disponibles para estudiantes, instituciones académicas, profesionales y empresas. Encuentre la formación de BIOVIA adecuada para usted.
Obtener ayuda
Encuentre información sobre certificación de software y hardware, descargas de software, documentación del usuario, contacto con soporte y oferta de servicios