
Structure-based Design
Comprehensive, Scalable Portfolio of Scientific Tools to Support Structure-based and Fragment-based Design
Facilitate Drug Discovery From Hit Discovery to Late-stage Optimization
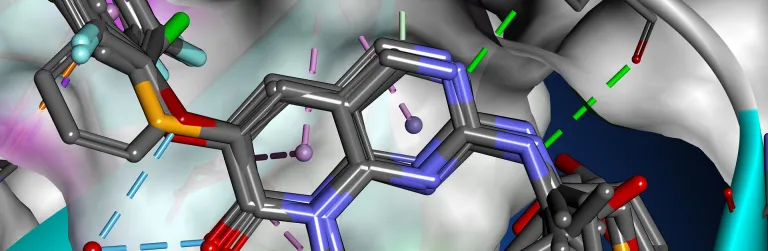
Structure-Based Design (SBD) and the related Fragment-Based Design (FBD) are well established strategies in the rational development of small molecule drugs. Knowledge of how a small molecule binds into a protein affords considerable advantages, both in terms of prioritizing compounds for early stage screening, through to optimizing potency and selectivity.
BIOVIA Discovery Studio delivers a comprehensive, scalable portfolio of scientific tools, including GOLD from the Cambridge Crystallographic Data Centre (CCDC), to support and assist SBD and FBD strategies from hit discovery to late-stage lead optimization.
- Prepare
- Screen
- Score
- Extend
Prepare
- Analyze and prepare 3D structures (e.g., PDB, X-ray structure, homology model) for SBD
- Automatically build neighboring molecules based on crystal packing and analyze their interactions
- Predict residue ionization states at chosen pH
- Identify and study putative ligand binding sites
- Prepare ligands with extensive set of characteristics and calculate 3D coordinates
- Generate ligand conformations
- Filter ligands based on drug-likeness, molecular properties, or to remove undesirable groups or features
Screen
- Hit Identification and optimization
- Perform virtual screening on ligands and fragments using either the CATALYST pharmacophore engine, or the LibDock or CDOCKER docking approaches
- Perform docking with GOLD §
- Perform in situ lead optimization using classical medicinal chemistry reaction transformations and commercially-available reagents
- Scaffold-hop or perform R-group substitutions in situ using molecular fragments derived from commercially-available compounds
§ Requires license from Cambridge Crystallographic Data Centre
Score
- Calculate binding energies with MM-PBSA or MM-GBSA CHARMm-based methods
- Accurately predict relative ligand binding energy for a congeneric ligand series using the free energy perturbation (FEP) method
- Calculate the relative free energy of binding for a combinatorial library of ligands modeled by Multi-Site Lambda Dynamics (MSLD)
- Identify critical interacting residues using a comprehensive set of favorable, unfavorable and unsatisfied non-bond monitors
- Profile and prioritize screening hits, optimizing potency and target specificity
Extend
- Design and optimize combinatorial libraries as new starting points for further screening.
- Combine your scores with classical QSAR, fingerprints, and Quantum Mechanics based descriptors and create advanced predictive models
- Minimize toxicity using TOPKAT and optimize the pharmacokinetic profile.
Start Your Journey
Accelerate your drug discovery with BIOVIA Discovery Studio.
Join the conversation in the BIOVIA Drug Discovery & Development Community!
Also Discover
Comprehensive Portfolio of Validated Scientific Tools for Every Aspect of Macromolecule-based Research
A Rich Set of In Silico Tools to Support The Design of Biotherapeutics

Protocols for State-of-the-art Molecular Dynamics Simulations
De Novo Drug Design, Multi-Target Drug Design and Activity Profiling
Tools for Designing Therapeutics with Favorable Pharmacokinetic Properties and Safety Profiles
Free, Feature-Rich Molecular Visualizer
Comprehensive Modeling and Simulation for Life Sciences Research

Accelerate Innovation in Life Sciences Research and Development
Learn What BIOVIA Can Do for You
Speak with a BIOVIA expert to learn how our solutions enable seamless collaboration and sustainable innovation at organizations of every size.
Get Started
Courses and classes are available for students, academia, professionals and companies. Find the right BIOVIA training for you.
Get Help
Find information on software & hardware certification, software downloads, user documentation, support contact and services offering