
BIOVIA COSMOtherm
Advanced COSMO-RS Implementation with Quantum Chemically Generated Charge Density Surfaces
Predictive Thermodynamics for Liquids
BIOVIA COSMOtherm is the universal tool for predictive property calculation of liquids, and combines quantum chemistry and thermodynamics in a unique fashion. It calculates the chemical potential of almost any molecule in almost any pure or mixed liquid at variable temperature, i.e. it predicts how happy a molecule is in a certain liquid environment. This is the key for the prediction of a multitude of properties required in industrial applications or academic research, including solubility, partitioning, vapor pressure, and complete phase diagrams.
In contrast to several other available methods BIOVIA COSMOtherm is able to predict properties as function of concentration and temperature by applying thermodynamically consistent equations.
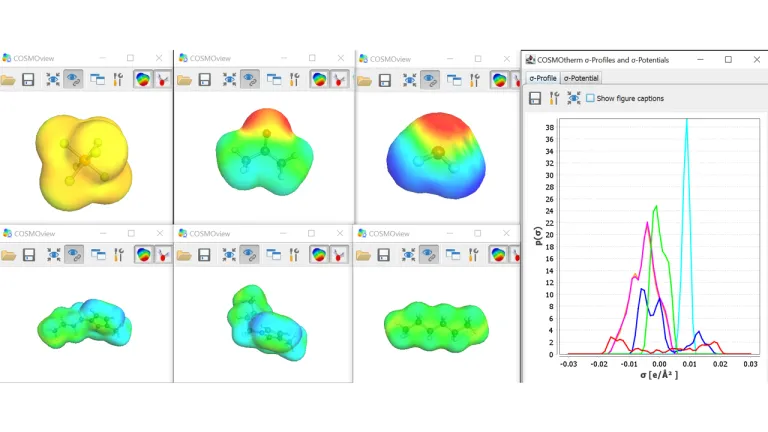
Features
- Predictable Properties
- Ready out of Box
- Fewer Complications
- Application Range
Predictable Properties
- Solubility of liquids, solids and gases
- Activity coefficients, two-phase partitioning (e.g. LogP), liquid extraction
- Conformer relevance in different mixtures
- Phase behavior: liquid-liquid, liquid-vapor and liquid-solid equilibria
- Vapor pressures, free energy of solvation, Henry's law constant
- pKa, dependence of chemical reaction equilibria on the solvent
- Energy of transfer to a flat liquid-liquid interface
- Micelle and membrane partitioning
- Interfacial tension, adsorption and environmental properties
Ready out of the box
BIOVIA COSMOtherm features an easy to use graphical interface to allow users to focus more on research and less on the software. In addition, the completely input file-oriented command line version allows seamless integration into existing workflows or batch processing. The greatest advantages of BIOVIA COSMOtherm are its general applicability and its high predictivity. Once integrated into the workflow, it can be applied to almost the complete range of organic chemistry without needing a scientist to think about missing parameters or applicability for a specific case.
Fewer Complications
- No missing parameters as in force-field simulations, UNIFAC or equations of state
- No case-specific choices of any kind
- Predictivity far beyond fitting sets
- Multifunctional and complex compounds are welcome
- Results within seconds or minutes
- (a time-consuming DFT calculation has to be conducted for each molecule once, the results are then stored and can be reused for all properties and mixtures later on)
Application Range
- Limited to incompressible liquids, sufficiently below the critical point
- Gas phase chemical potential is only valid for ideal gases
- Polymers can only be predicted as solvents for small molecules
- Long-range interactions of ions are missing (e. g. Debye-Hückel term)
Start Your Journey
The world of COSMO chemistry is changing. Discover how to stay a step ahead with BIOVIA.
Also Discover

The Shortcut to COSMO-RS

Efficient Treatment of Self-organizing Systems and Biomembranes

Foster Innovation with In Silico Design
Learn What BIOVIA Can Do for You
Speak with a BIOVIA expert to learn how our solutions enable seamless collaboration and sustainable innovation at organizations of every size.
Get Started
Courses and classes are available for students, academia, professionals and companies. Find the right BIOVIA training for you.
Get Help
Find information on software & hardware certification, software downloads, user documentation, support contact and services offering